1. ユビキチン化翻訳後修飾
細胞内のタンパク質分解系は主に、オートファジー-リソソーム系、カルパイン系、ユビキチンを介在するプロテアソーム分解系などが知られている。
ユビキチン介在性タンパク質分解系による発現調節は、1)生化学的には不可逆的反応であることと、2)迅速に、標的となるタンパク質質を認識し分解するシステムということに特徴づけられる。 最近では、タンパク質分解に関係しないユビキチン化も知られている(キナーゼの活性化、エンドサイトーシス、オートファジー、DNA修復、ヒストン制御など)。
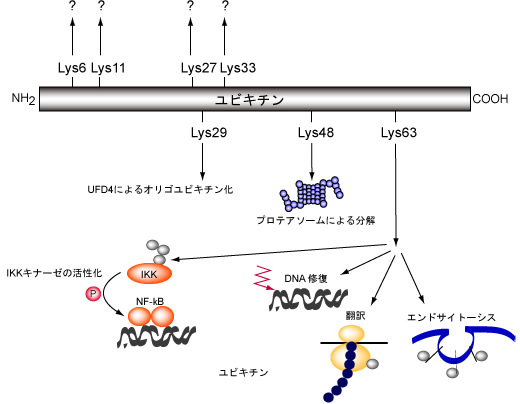
図1: ユビキチン・プロテアソームシステムが制御する細胞内高次機能
ユビキチン化は機能的に多くの細胞内現象にかかわり、特に細胞内シグナル伝達、細胞周期、細胞膜受容体介在性エンドサイトーシス、転写、オルガネラ合成、精子形成調節などに関与している。ユビキチンが介在するプロテアソーム分解系の生化学的反応についての研究は、最近めざましい発展がみられ、ユビキチン活性化酵素(E1)、ユビキチン結合酵素(E2)、ユビキチンタンパク質リガーゼ(E3)が分子クローニングされ、さまざまな標的タンパク質において特異的なユビキチン化に関与する酵素が明らかにされている。 ユビキチン化された標的タンパク質質はプロテアソームの19Sキャップに存在するRpn10/S5aという分子に認識され、20Sを構成するa7b7サブユニットで分解を受ける。
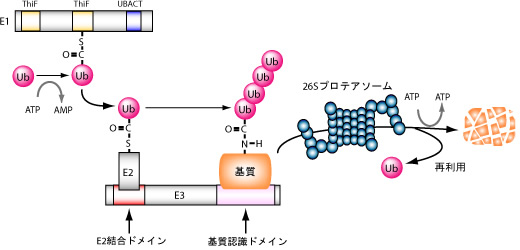
図2: ユビキチン化反応の分子論
ユビキチン化反応は,E1(ユビキチン活性化酵素),E2(ユビキチン結合酵素),E3(ユビキチンリガーゼ)から構成されるカスケードによって触媒されるタンパク質の翻訳後修飾機構である.ポリユビキチン化されたターゲットタンパク質はプロテアソームに認識され分解を受ける.
2. ユビキチンリガーゼE3の多様性
分子レベルでユビキチン化の解析が進んだものとして、ヒトパピローマウイルス遺伝子産物E6と結合し癌抑制遺伝子であるp53を分解するE3としてE6AP(E6-associated protein)というユビキチンリガーゼが同定されている。 DNAの損傷をモニターする転写因子として重要な癌抑制遺伝子産物p53はヒトパピローマウイルスが感染した細胞(特に子宮頸部扁平上皮細胞)では、その産物のひとつであるE6が物理的にp53と生体内のE6APと3量体を形成し、ユビキチンリガーゼ活性を有するE6APがp53をユビキチン化に導くということが報告された。E6APはユビキチンとチオエステル結合する活性化システイン残基をそのC末端側に有し、その発見はユビキチン化がE1-E2-E3というユビキチン化転移反応を分子レベルで存在することを証明するものとなった。さらに、そのユビキチン結合部位を含む約300アミノ酸が、さまざまな真核細胞の多くのタンパク質に広く保存された配列であることが判明し、HECTドメイン(homologous to E6AP carboxy terminus)と呼ばれ、このドメインを有するタンパク質はユビキチンリガーゼとして機能することが認められている。現在まで、E6AP以外にNedd4、Rsp5、Pub1、Tom1、Ufd4、Smurf1/2、p100などが同定されている。
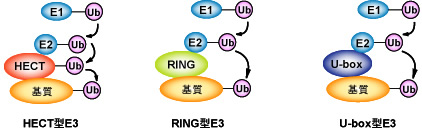
図3: 多様なユビキチンリガーゼ(E3)群
ユビキチンリガーゼ(E3)はユビキチン化の基質認識を司る酵素であり,現在ではドメイン構造の違いにより,HECT型,RING型,U-ボックス型の3種類のユビキチンリガーゼ群に分類される.HECT型E3は自分自身もシステイン残基にユビキチンとチオエステル結合する中間体を形成する.
CblはPDGF受容体やEGF受容体などのタンパク質としての安定性に関与することが示されていた。実際に、Cblには以前からRINGフィンガードメイン(really interesting new gene)と呼ばれる亜鉛と結合するモジュールが存在し、その部分が標的タンパク質の安定性に関与することが示されていた。 時を同じくして、UbcH5BというヒトE2をbaitにしたyeast two-hybridスクリーニングにより、RINGフィンガーを有した分子(AO7)が同定され、RINGフィンガードメインを有した分子にユビキチンリガーゼ活性が認められることが確認された。
NF-kBシグナルの抑制系調節分子IkBやWnt/Winglessシグナルの介在分子b-カテニンのユビキチン化介在性分解を司る分子複合体型ユビキチンリガーゼとして、SCF(Skp1/Cul1/F-box protein)複合体が同定され(SCFFbw1)、その後、細胞周期のG1/S期の移行に重要なCDKインヒビターp27やサイクリンEのユビキチン化に作用するユビキチンリガーゼとして、SCFSkp2が同定された。さらにSCFの第4の構成成分として、Rbx1/ROC1が同定され、この分子にはRINGフィンガーモチーフが存在することが示された。さらに、M期サイクリンであるサイクリンBのユビキチン化を遂行するAPC/C (anaphase promoting complex/ cyclosome)の構成分子が網羅的に同定され、そのなかにRINGフィンガードメインを有するAPC11が存在することが示された。上述の2つのRINGフィンガータンパク質は複合体として、ユビキチンリガーゼ活性を示すが、前述のようにCblをはじめ、Mdm2、IAP、Siah-1、BRCA1などのように単独でもユビキチンリガーゼとして働く分子も存在している。
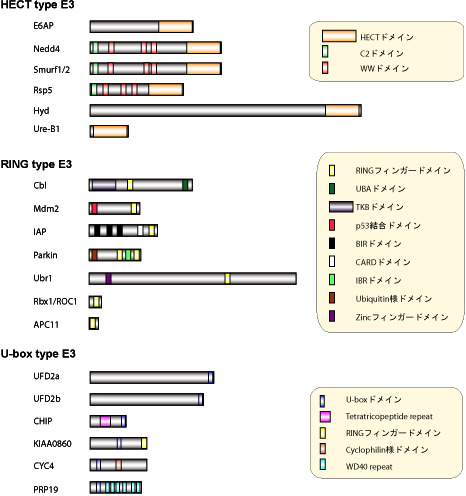
図4: 多様なユビキチンリガーゼ(E3)群の分子構造
ユビキチンとb-ガラクトシダーゼの融合タンパク質(Ub-bgal)の安定性を指標に、その分解を制御している分子の同定が、発芽酵母の遺伝学を使用したスクリーニングで行われた。そのユビキチン化反応系は、E1、E2(Ubc4)、及びUFD4 (HECT型ユビキチンリガーゼ)、さらにUFD2とよばれたポリユビキチン鎖を伸長させる作用を示す分子(E4)によって遂行されていることが報告された。構造解析より、HECTドメインもRINGドメインもE2と結合する領域が似ていることが示されているが、このUFD2の“U-ボックス”とよばれる領域はタンパク質配列上、RINGフィンガーを類似していることがコンピュータデータベース解析より明らかとなっている。
3. TRIMファミリータンパク質の多様性と機能
TRIM(tripartite motif)タンパク質の多くはRINGドメインを有し、そのほとんどがE3ユビキチンリガーゼとして機能することが報告されている。TRIMタンパク質はヒトやマウスにおいては70以上の遺伝子が存在し、多くのものは各染色体に遺伝子クラスターを形成して存在している(ヒトの場合、第12、18、20、21、22番及びY染色体には存在しない)。TRIMタンパク質は、アミノ末端側にRINGドメイン、Bボックスドメイン、コイルドコイルドメインを有し、カルボキシル末端側にさまざまなドメイン構造(SPRY、FN3、PHD、BROMO、NHL、MATH、ARF、EXOIIIなど)を有し、そのカルボキシル末端側の構造多様性から11種のサブファミリーに分類される。
TRIMタンパク質の祖先遺伝子は原生生物で既に存在しており、線形動物(線虫など)、節足動物(ショウジョウバエなど)にも存在するが、SPRYドメインを有するTRIMタンパク質は脊椎動物において増加している。これまでにシロイヌナズナのゲノムに完全なTRIMタンパク質の遺伝子は見つかっていないが、RINGドメインを持たないがBボックスドメインを有した類似構造のタンパク質が同定されている。TRIMタンパク質はホモダイマーやヘテロダイマーを形成することが知られている。よって、ダイマーのパートナーを入れ替えることにより、基質特異性に関して多様性の増加をもたらす可能性がある。
TRIMタンパク質が使用するE2の網羅的解析により、多くがクラスDもしくはクラスEのE2(E2D1、E2D2、E2D3、E2E1、E2E2及びE2E3)を使用することが示されている。また、いくつかのTRIMタンパク質はE2N (Ubc13)と相互作用し、リジン63型ポリユビキチン鎖を形成することが推測されている。また、ある種のTRIMタンパク質はsmall ubiquitin-related modifier-1(SUMO-1)のE2であるUbc9(E2I)と相互作用し、標的分子のSUMO-1化を促進することが示されている。このように、TRIMタンパク質はタンパク質分解のためのE3ユビキチンリガーゼのみならず、非定型的なユビキチン化やSUMO化などのE3リガーゼとして働く可能性がある。
TRIM23
脂肪組織は余剰エネルギーを貯蔵する主要組織であるが、近年の研究により、エネルギー代謝制御ホルモンを産生する内分泌組織としての役割が注目されている。脂肪細胞の分化は種々の転写因子が適切な順番で発現することで進行するが、ペルオキシゾーム増殖剤応答性受容体ガンマー(PPARg)は脂肪細胞分化に必須であり、かつその過剰発現により脂肪細胞への分化誘導が可能なマスター遺伝子として機能する。脂肪細胞分化が始まると、まずC/EBPb、C/EBPdなどの初期転写因子の発現が誘導され、これら初期転写因子はPPARg、C/EBPaなどの後期転写因子の発現を誘導する。PPARgとC/EBPaは互いの発現を誘導し合うと同時に、協調して脂肪細胞特異的な遺伝子の発現を誘導し、成熟脂肪細胞へと分化させる。
我々はTRIM23をノックダウンさせた場合、白色脂肪前駆細胞3T3-L1では脂肪細胞への分化成熟およびPPARgの発現誘導が抑制されることを見出した。TRIM23をノックダウンさせた細胞において分化誘導時のPPARgプロモーターの変化を調べたところ、転写因子C/EBPb、C/EBPdの結合やエピジェネティックなクロマチン修飾の変化、オープンクロマチン構造の変化は変化がなかったが、PPARgの安定性が減少することが判明した。さらに、TRIM23はPPARgに非定型的なポリユビキチン鎖を形成することで、プロテアソームによる認識及び分解を抑制することがわかった。最近、脱分化脂肪細胞株を使用した実験により、癌幹細胞のアクチンファイバーを脱重合させることで終末分化させることができる可能性も示唆されており、TRIM23を活性化させる方法が開発できれば、抗癌剤のシーズに使えるかもしれない。
TRIM29
癌細胞は上皮間葉転換 (epithelial to mesenchymal transition: EMT) を経て、転移する能力を獲得すると想定されている。EMTを経た癌細胞は、細胞外マトリックスへの接着能や浸潤能に変化を示す。EMTに関わる遺伝子の発現は、SNAIL1/2、TWIST1/2、ZEB1/2などの転写因子によって制御されることが知られている。p53遺伝子ファミリーの一つであるp63も、EMTを抑制する機能を有し、上皮細胞分化のマスターレギュレーターとして同定されている。p63遺伝子には2つの異なるプロモーターがあり、transactivating p63 (TAp63) 及びN-terminal truncated p63 (DNp63) のアイソフォームが発現される。TAp63は癌細胞の転移を抑制する機能を有するが、DNp63はTAp63に対してドミナントネガティブな作用を示すことが知られている。
TRIM29は常染色体劣性遺伝性疾患である毛細血管拡張性運動失調症 (ataxia telangiectasia: AT) の機能欠損を補う遺伝子として、ヒトのコスミドライブラリーを用いた機能相補実験によって同定された。TRIM29は一般的なRINGフィンガードメインを持たず、C末端領域においても特定のドメイン構造が保存されていない。ATは神経変性、免疫不全、早老や高発癌などの多彩な表現型を有する疾患であり、その原因遺伝子としてATM (ataxia telangiectasia mutated) が同定されている。AT細胞の機能相補実験の結果と一致して、TRIM29はSiHa細胞、NIH 3T3細胞やBxPC3細胞において放射線耐性能の制御に関与することが報告されている。
TRIM29はp63と同様に、正常扁平上皮細胞に特異的に発現しており、病理学的解析から膀胱癌、結腸直腸癌、胃癌、肺癌及び膵臓癌などの悪性度と密接に関係していることも報告されている。我々は、乳癌細胞において、TRIM29はTWIST1の発現を抑制することによって浸潤能を抑制していることを報告した。また、我々はTRIM29が前立腺組織におけるバイオマーカーとして機能する可能性を見出した。正常な前立腺は、腺細胞及び基底細胞からなっており、腺細胞の周囲に基底細胞が存在する組織像が観察される。そして、前立腺癌において、基底細胞は消失することが知られている。我々は正常前立腺組織の基底細胞にTRIM29が特異的に発現していることを見出した。正常前立腺組織と前立腺癌組織を使った抗TRIM29抗体による免疫組織化学染色により、前立腺癌組織においてTRIM29陽性基底細胞が消失することが判明し、前立腺癌におけるTRIM29の診断バイオマーカーとしての有効性を示されている。また、我々はTRIM29結合タンパク質としてアセチル化酵素TIP60を同定し、TRIM29の過剰発現はTIP60の分解を促進し、TIP60によるp53のアセチル化を抑制すること、大腸癌細胞株において紫外線照射による細胞死を抑制することを示した。以上より、TRIM29は癌化を促進する分子として機能する可能性が示唆されている。
我々はさまざまなヒト癌細胞株におけるTRIM29の発現レベルを検討したところ、子宮頸癌細胞株(HeLaやSiHaなど)に発現が高いことが判明した。そこで我々は、TRIM29による子宮頸癌細胞における機能変化を検討した。TRIM29の過剰発現もしくはノックダウンにより、細胞接着能および浸潤能が変化することが明らかになったが、子宮頸癌細胞株によりTRIM29による反応性は異なっていた。また、TRIM29ノックダウンによってインテグリンの発現パターンが変化し、ZEB1の発現レベルが上昇した。さらに、遺伝子発現制御におけるTRIM29の機能を予測するために、TRIM29の結合タンパク質の同定を質量分析計およびプルダウンアッセイ法を用いて行った。すると、TRIM29はTAp63、RNAポリメラーゼII及びMediator複合体に結合することが判明した。また、TRIM29をノックダウンした細胞では、ZEB1およびITGB1の発現レベルが上昇していた。ただし、TRIM29によるZEB1及びITGB1の発現上昇は、TAp63の過剰発現によって抑制されなかった。すなわち、TRIM29はTAp63非依存的にZEB1およびITGB1の発現を制御していることが示唆された。以上より、EMTや癌細胞の制御におけるTRIM29のさらなる機能解明は、癌の新たな診断や治療方法の開発に重要かもしれない。
DNA修復過程に関与するTRIM29
真核生物において、ゲノムDNAはクロマチン構造として細胞核内に折りたたまれており、クロマチン構造はヌクレオソームを基本単位として構成されている。ヌクレオソームは、4種類のヒストンコアタンパク質H2A、H2B、H3及びH4をそれぞれ2分子ずつ含むヒストンオクタマーにDNAが左巻きに巻き付くことによって形成される。ヒストンのN末端領域はヒストンテールと呼ばれ、さまざまな翻訳後修飾を受けることによって、多様な機能に関与する。ヒストン翻訳後修飾として、リジン残基のメチル化、アルギニン残基のメチル化、リジン残基のアセチル化、セリン残基のリン酸化、リジン残基のユビキチン化などが知られ、翻訳後修飾を受けたヒストンはDNA修復タンパク質、転写調節因子、クロマチン再構成因子などの足場として機能する。
ゲノムDNAの安定性維持は生命活動を正常に行うために重要であるが、DNAは外的および内的要因によって日常的に損傷を受けている。DNA損傷の中でも、DNA二本鎖切断 (DNA double strand break: DSB) は、生物にとって危険度の高いDNA損傷であり、電離放射線 (ionizing radiation: IR) やアルキル化剤などによって起きる。そして、DSB修復能力の欠損や低下は、癌や免疫疾患などのさまざまな疾患を起こす。生物はDSBを感知し、DSB部位にDNA修復タンパク質を集積させるDNA修復機構を有している。DNA損傷応答 (DNA damage response: DDR) の初期に、MRE11-RAD50-NBS1 (MRN) 複合体とKu70-Ku80複合体がDSB断端に結合し、ATMとDNA-PKcsなどのプロテインキナーゼが活性化され、ヒストンH2AXの139番目のセリン残基がリン酸化される(gH2AX)。gH2AXはクロマチン上でfociと呼ばれる集合体を形成し、DNA修復タンパク質はgH2AXを足場にしてDSB部位に集積し、相同組換え (homologous recombination: HR) もしくは非相同末端接合 (nonhomologous end-joining: NHEJ) によるDSB修復を促進する。DSBに応答したH4のアセチル化は、ヒストンアセチルトランスフェラーゼTIP60によって選択的に行われ、さらにTIP60がATMをアセチル化することによって、ATMを活性化させる。DDR初期において機能するタンパク質に加えて、BRCA1、コヒーシンやDNAミスマッチ修復タンパク質などのタンパク質もDSB損傷部位に蓄積し、DSB修復に関わることが報告されている。DNAミスマッチ修復 (DNA mismatch repair: MMR) タンパク質は、クロマチン上でATMと複合体を形成し、DNA損傷に応答する。E3ユビキチンリガーゼBRCA1は、ATM、MRN複合体やMMRなどとBRCA1-associated surveillance complex (BASC) と呼ばれる複合体を形成する。
最近、DSB修復に関与するE3ユビキチンリガーゼとして、RNF8とRNF168が報告されている。ATMによってリン酸化されたMDC1にRNF8が結合することで、RNF8のよるリジン63型ポリユビキチン鎖を形成がが促進され、53BP1、RAP80/BRCA1をDSB部位に集積される。さらに、ATMによってリン酸化されたHERC2を呼び寄せることでRNF8-UBE2N(Ubc13)の結合安定性を増加し、さらにはリジン63型ポリユビキチン鎖形成の増強を図ることが報告されている。ただし、この現象はニワトリ由来B細胞株であるDT40を使用した実験では証明されていない。RNF168はユビキチン結合ドメイン(MIUドメインとUMIドメイン)を有しており、RNF8によって形成されたユビキチン鎖に結合する。その後、RNF168はヒストンH2Aのリジン13/15をユビキチン化することで、DSB部位に53BP1、RAP80/BRCA1を集積させる。このユビキチン化に関して、USP3、USP16、USP44、OTUB1などの脱ユビキチン化やRNF169などが抑制的に働くことが報告されている。このようにDNA修復メカニズムが正常に活性化するためには、DNA修復タンパク質による複雑な化学反応(リン酸化やユビキチン化)や分子間相互反応が必要とされる。
我々はTRIM29が放射線耐性能に関わることから、TRIM29はDNA修復タンパク質の機能を制御しているのではないかと推測した。まず、TRIM29の細胞内局在を解析したところ、子宮頸癌細胞株HeLaにおいてTRIM29はクロマチン上に存在することが明らかとなった。次にTRIM29のクロマチン上での機能を予測するために、質量分析解析によるTRIM29の結合タンパク質の同定を行った。その結果、TRIM29はBASCなどのDNA修復タンパク質と複合体を形成し、DNAミスマッチ修復タンパク質のひとつであるMSH2と直接結合することが判明した。TRIM29をノックダウンした細胞では、DNA修復開始シグナルであるgH2AXおよびH4K16Acの低下が観察された。また、TRIM29はヒストンに直接結合し、H3K36me3を認識することが明らかとなった。そして、TRIM29のクロマチンへの結合は、gH2AXの誘導および放射線耐性能の維持に必要であることが判明した。以上の結果から、TRIM29はH3K36me3を介してクロマチンに結合し、DNA修復タンパク質をクロマチンへ集積させることによって、ゲノム安定性の維持に寄与していることが明らかになった。TRIM29のクロマチンへの結合は、ATMやDNA-PKcsの活性化に関わることが予想されるが、TRIM29によるATMやDNA-PKcsの活性化メカニズムについては未だ不明である。今後は、TRIM29によるDNA修復タンパク質のクロマチン上への集積及びDNA修復経路の活性化の制御機構についてさらなる検討を行う必要があると思われる。
参考文献:
1. Hatakeyama, S. (2017) TRIM family proteins: roles in autophagy, immunity, and carcinogenesis.
Trends Biochem. Sci. 42, 296-310.
2. Watanabe, M & Hatakeyama, S. (2017) TRIM proteins and diseases.
J. Biochem. 161, 135-144.
3. Hatakeyama, S. (2011) TRIM proteins and cancer.
Nat. Rev. Cancer 11, 792-804.
4. Watanabe, M., Takahashi, H., Saeki, Y., Ozaki, T., Itoh, S., Suzuki, M., Mizushima, W., Tanaka, K. and Hatakeyama, S. (2015) The E3 ubiquitin ligase TRIM23 regulates adipocyte differentiation via stabilization of the adipogenic activator PPARgamma.
eLife 4, e05615.
5. Masuda, Y., Takahashi, H. and Hatakeyama, S. (2015) TRIM29 regulates the p63-mediated pathway in cervical cancer cells.
Biochim. Biophys. Acta 1853, 2296-2305.
6. Masuda, Y., Takahashi, H., Sato, S., Tomomori-Sato, C., Saraf, A., Washburn, M.P., Florens, L., Conaway, R.C., Conaway, J.W. and Hatakeyama, S. (2015) TRIM29 regulates the assembly of DNA repair proteins into damaged chromatin.
Nat. Commun. 6, 7299.
- 北海道大学 大学院医学研究院生理系部門
生化学分野 医化学教室 - 〒060-8638 札幌市北区北15条西7丁目
医学部 北研究棟 1階N1-111
TEL: 011-706-5047
FAX: 011-706-5169